Molekularno genetsko testiranje je pomembno orodje za postavitev diagnoze spinalne mišične atrofije6, 7
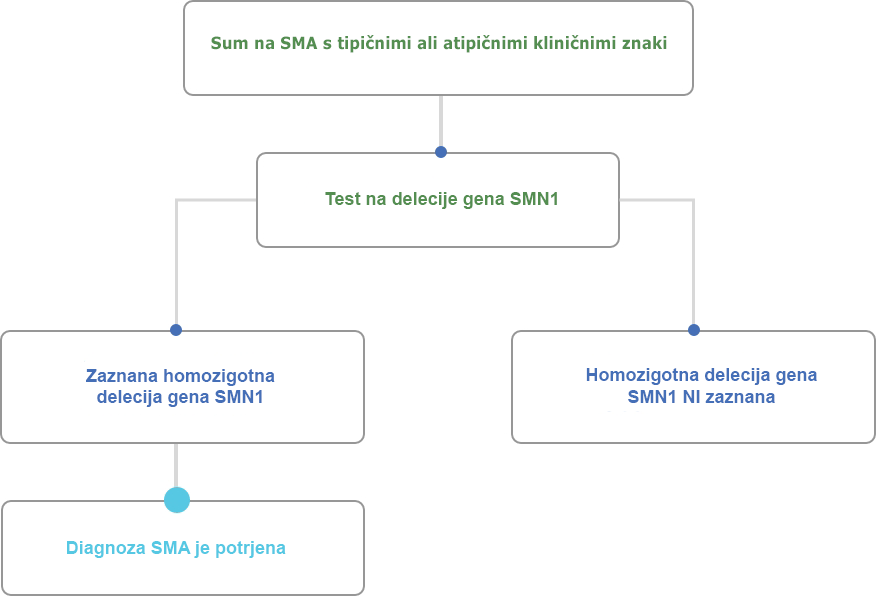
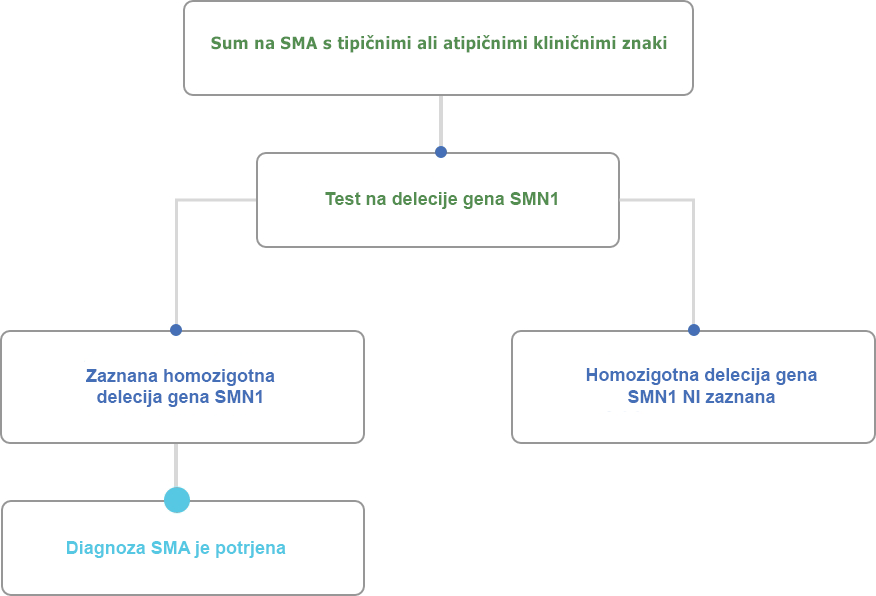
Povzeto po D`Amico et al.6
SMA je rezultat homozigotnih delecij ali mutacij, ki vključujejo gen SMN v lokusu 5q13 kromosoma 5. Obstaja veliko redkih živčno-mišičnih bolezni (npr. Lambert-Eaton-ov miastenični sindrom, ki prizadene 0,05 - 0,08 / 100.000 ljudi na leto).8 Te bolezni lahko vključujejo mutacije različnih genov, ki niso povezani s 5q13.
Diferencialna diagnoza 5q spinalne mišične atrofije vključuje, ni pa omejena na:9
Bolezni hrbtenjače
- Neoplazme
- Ostale mielopatije
Miopatije
Prirojene miopatije:
- Prirojena miotonična distrofija
- Prirojene mišične distrofije
- Mišične distrofije
- Mitohondrijske miopatije
- Pompejeva bolezen
Ostale metabolne miopatije:
- Vnetne miopatije
- Kanalne miopatije
Nevropatije
- Prirojena hipomielinirajoča ali aksonska nevropatija
- Dedne motorične ali senzorične nevropatije
- Kronična vnetna demielinizirajoča polinevropatija
Bolezni živčno-mišičnih stikov
- Botulizem
- Prirojeni miastenični sindrom
- Lambert-Eatonov miastenični sindrom
- Avtoimunska miastenija gravis
Ostale bolezni motoričnega nevrona
- Spinalna mišična atrofija z dihalno stisko (SMARD)
- Juvenilna mišična atrofija distalne zgornje okončine (bolezen Hirayama)
- Fazio-Londejeva bolezen
- Brown-Vialetto-van Laere sindrom
- Juvenilna amiotrofična lateralna skleroza
- Ostale ne 5q SMA
Ostale bolezni
- Kromosomske nepravilnosti
- Prader-Willijev sindrom
- Abnormalnosti centralnega živčnega sistema
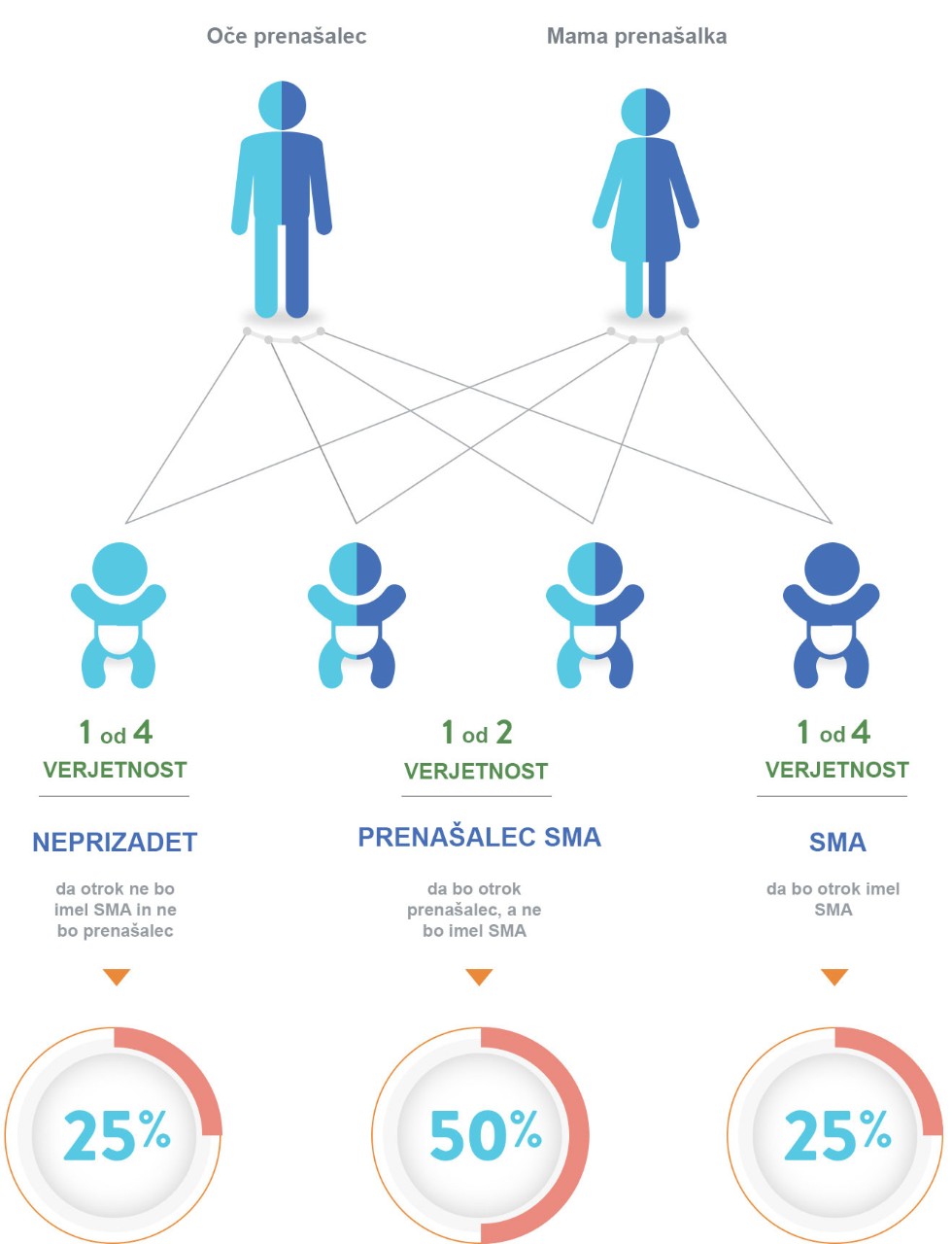
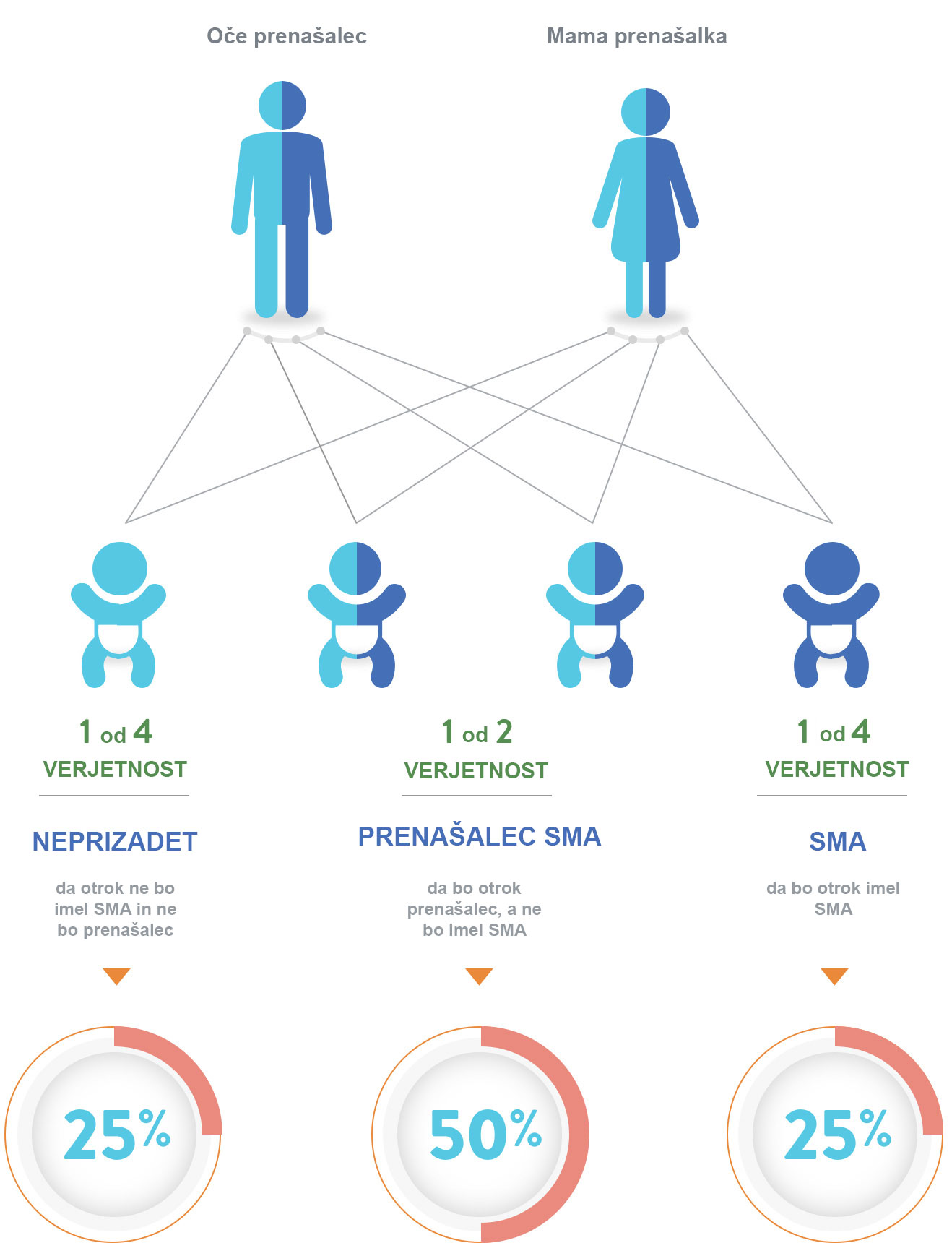
Gensko obolenje
Otrok podeduje dva izbrisana ali mutirana gena SMN1 -
po enega od vsakega starša4
Pomen zgodnje postavitve diagnoze
Naravni potek SMA vključuje nepopravljivo izgubo motoričnih funkcij.8 Po sicer začetnem povečanju, začetek SMA označujeta vrh motoričnih sposobnosti in poznejši upad, pri čemer je napredovanje bolezni (in izguba motoričnih sposobnosti) najhitrejše v zgodnji fazi.19
Zgodnja postavitev diagnoze je lahko pomemben dejavnik pri zdravljenju SMA.20
![]() Vzorec izgube motoričnega nevrona, opažen pri SMA, kaže na to, da je treba zdravljenje pri infantilnem tipu SMA (tip 1) začeti čim prej, vključno že v pre-simptomatskim obdobju pred znatno izgubo motoričnih nevronov.20
Vzorec izgube motoričnega nevrona, opažen pri SMA, kaže na to, da je treba zdravljenje pri infantilnem tipu SMA (tip 1) začeti čim prej, vključno že v pre-simptomatskim obdobju pred znatno izgubo motoričnih nevronov.20
![]() Molekularno genetsko testiranje je pomembno orodje za postavitev diagnoze SMA.7, 21
Molekularno genetsko testiranje je pomembno orodje za postavitev diagnoze SMA.7, 21
Ocena oblike SMA
Za oceno naravnega poteka bolezni in odziva na zdravljenje je bilo razvitih več lestvic za oceno motoričnih funkcij.22-24


