Spinalna mišična atrofija (SMA) je redka, dedna, avtosomno recesivna živčno-mišična bolezen, za katero je značilna degeneracija alfa motoričnih nevronov v sprednjih rogovih hrbtenjače, kar povzroči klinično progresivno atrofijo mišic, mišično oslabelost in izgubo gibljivosti. Najpogostejša oblika te bolezni se imenuje 5q SMA in predstavlja približno 95 % primerov.1,2
Trenutno SMA predstavlja eno najpogostejših in najresnejših genetskih bolezni v otroški dobi. Za to bolezen je značilno, da imajo otroci izrazito šibkost zgornjih in spodnjih okončin, lahko tudi šibke bulbarne in dihalne mišice, kognitivna sposobnost teh bolnikov pa se ne zdi prizadeta.2–6
Trenutno so bolniki s SMA razdeljeni na pet kliničnih podkategorij (tip 0, I, II, III in IV), ki so opredeljene glede na starost pojava simptomov bolezni, resnost bolezni in največjih doseženih gibalnih sposobnosti. Pri tipu 0 opazimo klinične znake že ob rojstvu in je najtežja oblika bolezni s pričakovano življenjsko dobo pod 6 mesecev.2,7
Ocena globalne prevalence SMA je približno 2–6 na 100.000 ljudi, ocena globalne incidence na 100.000 živorojenih pa od 3,5 do 7,1 za tip I, od 1,0 do 5,3 za tip II in od 1,5 do 4,6 za tip III.2,7,8 V Evropi se letna incidenca razlikuje od države do države in glede na fenotip.9
Genetska komponenta:
Leta 1995 je bilo ugotovljeno, da SMA povzroča homozigotna mutacija in/ali delecija gena na kromosomu 5q13, ki je danes znan kot gen za preživetje motoričnega nevrona 1 (Survival Motor Neuron – SMN1). Ta genetska sprememba je odgovorna za nezadostno proizvodnjo proteina SMN, ki je ključnega pomena za ohranjanje motoričnih nevronov, kar povzroči njihovo progresivno degeneracijo na ravni hrbtenjače.4,10,12
Vendar pa prisotnost drugega gena SMN (SMN2), ki je skoraj enak prvemu genu SMN, saj se od njega razlikuje le v 5 nukleotidih, omogoča nastajanje beljakovine SMN v manjših količinah. Pri tem nastane približno 90 % nestabilne beljakovine SMN, ki je nefunkcionalna in se hitro razgradi, in 10 % normalne beljakovine SMN.2,7,8,10,12
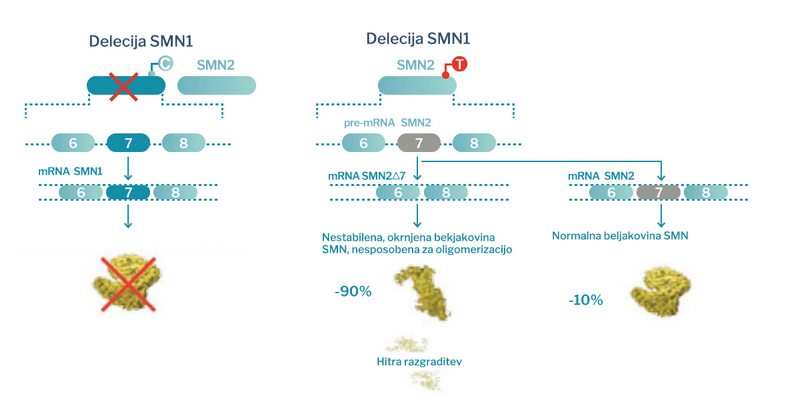
Slika 1. Normalno nastajanje beljakovine SMN prevedene iz gena SMN1 v primerjavi z alternativno potjo, nastajanjem iste beljakovine prevedene iz gena SMN2. Bolniki s SMA nimajo nobenega gena SMN1 in se zanašajo samo na alternativno pot nastajanja beljakovine SMN. pre-mRNA: predinformacijska ribonukleinska kislina; mRNA: informacijska ribonukleinska kislina
V tem smislu je treba poudariti, da je število kopij gena SMN2 sorazmerno z dobro prognozo in fenotipom SMA, tudi če proizvodnja funkcionalne beljakovine iz tega gena ne zadostuje za preživetje nevronov v osrednjem živčnem sistemu (OŽS).1,12